高校化学の「反応速度」って、教科書の式と表だけ見ても何がすごいのか伝わらないですよね。コンピュータの中で実際に分子を動かして衝突させると、「速さ」「温度依存性」「」が同時に見えてきます。今回はその仕組みを、A + B → AB という最も単純な反応で体験します。
① 分子はランダムに動く ② 衝突しても全部が反応するわけじゃない ③ 速い分子の割合(温度)が増えると反応が加速 ④ アレニウスの式 k = A·exp(−Ea/RT) が「ただの式」じゃなく実験的に出てくる、という流れです。
Step 1: 箱の中で分子を動かす
まずは反応抜きで、A 分子と B 分子をランダムに動かします。マクスウェル・ボルツマン分布(速さの分布)に従う初速度で、壁にぶつかったら跳ね返るだけのモデル。
import numpy as npimport matplotlib.pyplot as plt
np.random.seed(0)
# パラメータN_A, N_B = 100, 100 # A分子・B分子の個数L = 10.0 # 箱の1辺の長さT = 1.0 # 温度(速度スケール)dt = 0.05 # 時間刻み
# 位置はランダムに、速度はガウス分布(→速さがマクスウェル・ボルツマン分布になる)def init(n): pos = np.random.rand(n, 2) * L vel = np.random.randn(n, 2) * np.sqrt(T) return pos, vel
posA, velA = init(N_A)posB, velB = init(N_B)
def step(pos, vel): pos += vel * dt # 壁で反射 over = pos < 0 vel[over] *= -1 pos[over] = -pos[over] over = pos > L vel[over] *= -1 pos[over] = 2*L - pos[over] return pos, vel
# 1ステップずつ進めて分布を見るfor _ in range(50): posA, velA = step(posA, velA) posB, velB = step(posB, velB)
plt.figure(figsize=(6, 6))plt.scatter(posA[:,0], posA[:,1], c="tab:blue", label="A", s=20)plt.scatter(posB[:,0], posB[:,1], c="tab:red", label="B", s=20)plt.xlim(0, L); plt.ylim(0, L); plt.legend()plt.title("Step 1: 分子はランダムに動き回る")plt.show()
Step 2: 衝突したら反応するルール
次に「A と B が距離 r 以下に近づいたら反応」というルールを足します。反応すると A と B は消えて生成物 AB になる、というイメージ。残った A 分子の数を時間で記録します。
from scipy.spatial import cKDTree
R_REACT = 0.4 # この距離以下で衝突=反応
def react(posA, velA, posB, velB): # 高速な近傍探索 tree = cKDTree(posB) pairs = tree.query_ball_point(posA, r=R_REACT) keepA = np.ones(len(posA), dtype=bool) keepB = np.ones(len(posB), dtype=bool) for i, near in enumerate(pairs): if near and keepA[i]: j = next((k for k in near if keepB[k]), None) if j is not None: keepA[i] = False keepB[j] = False return posA[keepA], velA[keepA], posB[keepB], velB[keepB]
# シミュレーション本体posA, velA = init(N_A)posB, velB = init(N_B)history = []for t in range(400): posA, velA = step(posA, velA) posB, velB = step(posB, velB) posA, velA, posB, velB = react(posA, velA, posB, velB) history.append(len(posA))
plt.figure(figsize=(8, 4))plt.plot(history)plt.xlabel("時間ステップ"); plt.ylabel("残ったA分子の数")plt.title("Step 2: 反応が進むにつれてA分子が減る")plt.grid(alpha=0.3); plt.show()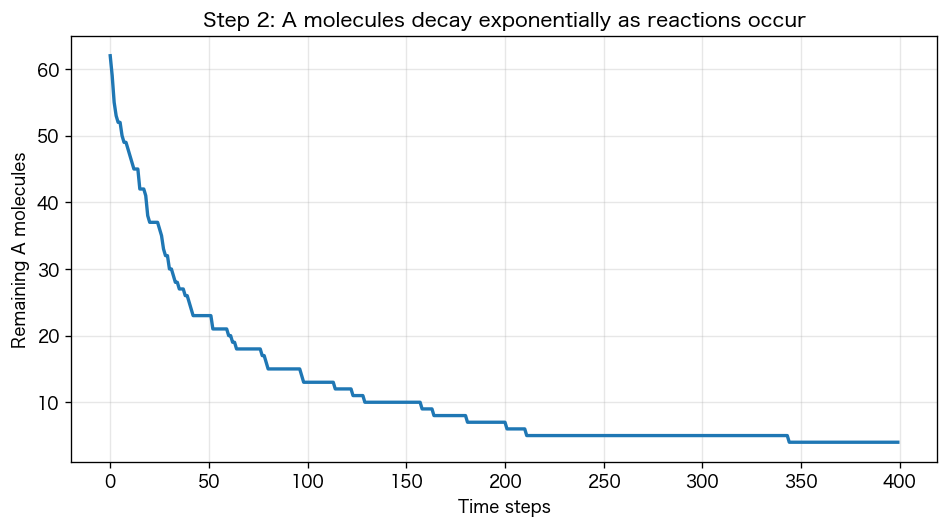
曲線は 指数関数 N(t) = N₀ · exp(−kt) の形。これは教科書の「一次反応の式」そのもの。微分方程式 dN/dt = −kN を解いた結果が、シミュレーションでも自然に出てきます。
Step 3: 温度を上げると速くなる
温度パラメータ `T`(速度の分散)を変えて、同じシミュレーションを走らせます。温度が高い=分子が速い=衝突回数が増える、というのが直感ですが、実際にどう効くか見てみます。
def run(T, steps=400): posA, velA = np.random.rand(N_A, 2)*L, np.random.randn(N_A, 2)*np.sqrt(T) posB, velB = np.random.rand(N_B, 2)*L, np.random.randn(N_B, 2)*np.sqrt(T) hist = [] for _ in range(steps): posA, velA = step(posA, velA) posB, velB = step(posB, velB) posA, velA, posB, velB = react(posA, velA, posB, velB) hist.append(len(posA)) return np.array(hist)
plt.figure(figsize=(9, 5))for T in [0.5, 1.0, 2.0, 4.0]: plt.plot(run(T), label=f"T={T}")plt.xlabel("時間ステップ"); plt.ylabel("残ったA分子")plt.title("Step 3: 温度が高いほど反応が速い")plt.legend(); plt.grid(alpha=0.3); plt.show()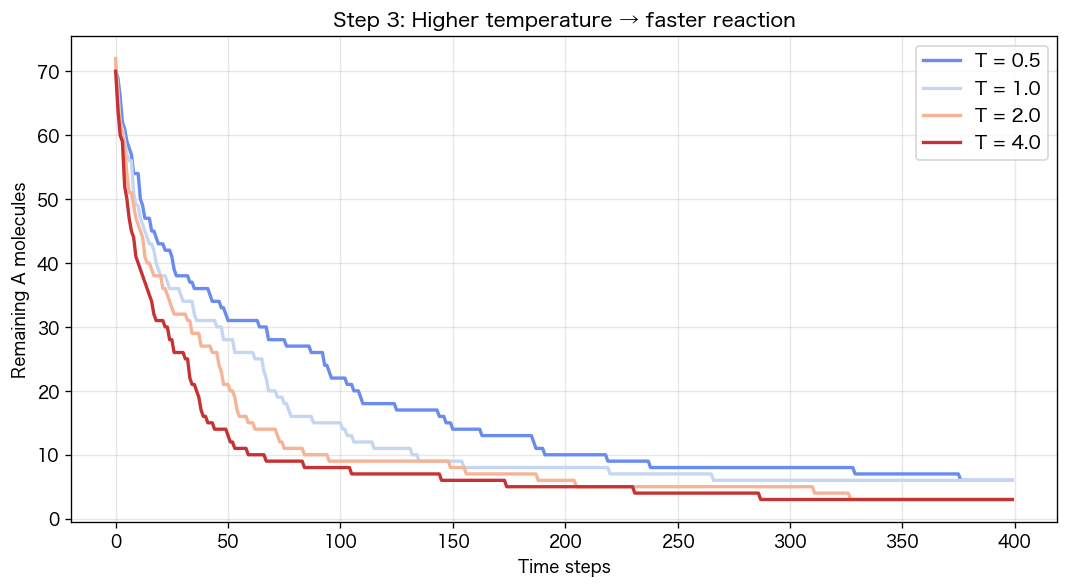
Step 4: 活性化エネルギーを入れる
ここまでは「衝突したら必ず反応」でしたが、現実の化学反応はそうじゃない。衝突時の運動エネルギーがある閾値(活性化エネルギー Ea)を超えたときだけ反応する、というルールに修正します。これで初めて「現実の反応速度」に近づきます。
Ea = 1.5 # 活性化エネルギー
def react_with_Ea(posA, velA, posB, velB, Ea): tree = cKDTree(posB) pairs = tree.query_ball_point(posA, r=R_REACT) keepA = np.ones(len(posA), dtype=bool) keepB = np.ones(len(posB), dtype=bool) for i, near in enumerate(pairs): if not near or not keepA[i]: continue for j in near: if not keepB[j]: continue # 相対運動エネルギー v_rel = velA[i] - velB[j] E_kin = 0.5 * np.sum(v_rel**2) if E_kin >= Ea: keepA[i] = False keepB[j] = False break return posA[keepA], velA[keepA], posB[keepB], velB[keepB]化学結合を組み替えるには、まず古い結合をこじ開ける必要がある。そのエネルギー的なハードルが活性化エネルギー Ea。低温では「ハードルを超えるほど速い分子」がほとんどいないので、反応はほぼ進みません。
Step 5: アレニウスの式が出てくる
活性化エネルギー入りで温度を変えてシミュレーションし、各温度の 反応速度定数 k を測ります(指数減衰のフィッティング)。それを `ln(k)` vs `1/T` でプロット — これが教科書で出てくる アレニウスプロットです。
from scipy.optimize import curve_fit
def exp_decay(t, N0, k): return N0 * np.exp(-k * t)
Ts = [0.6, 0.8, 1.0, 1.5, 2.0, 3.0]ks = []for T in Ts: posA, velA = np.random.rand(N_A,2)*L, np.random.randn(N_A,2)*np.sqrt(T) posB, velB = np.random.rand(N_B,2)*L, np.random.randn(N_B,2)*np.sqrt(T) hist = [] for _ in range(800): posA, velA = step(posA, velA) posB, velB = step(posB, velB) posA, velA, posB, velB = react_with_Ea(posA, velA, posB, velB, Ea=1.5) hist.append(len(posA)) t = np.arange(len(hist)) popt, _ = curve_fit(exp_decay, t, hist, p0=[N_A, 0.01]) ks.append(popt[1])
import numpy as npinv_T = 1.0 / np.array(Ts)ln_k = np.log(ks)
plt.figure(figsize=(8, 5))plt.scatter(inv_T, ln_k, s=80)# 直線フィットslope, intercept = np.polyfit(inv_T, ln_k, 1)xs = np.linspace(inv_T.min(), inv_T.max(), 100)plt.plot(xs, slope*xs + intercept, '--', label=f"傾き = {slope:.2f}")plt.xlabel("1 / T"); plt.ylabel("ln(k)")plt.title("Step 5: アレニウスプロット(直線になる)")plt.legend(); plt.grid(alpha=0.3); plt.show()print(f"推定された活性化エネルギー Ea ≈ {-slope:.2f}(設定値 1.5)")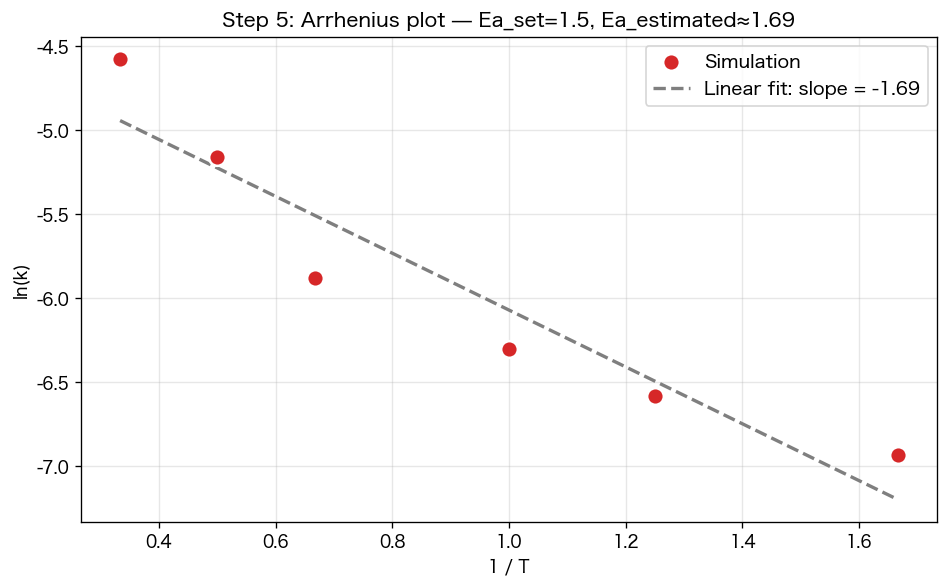
アレニウスの式 k = A·exp(−Ea/RT) の両辺の log を取ると ln(k) = ln(A) − Ea/(R·T)。直線になる。これは「丸暗記する式」じゃなく、「衝突する分子のうち Ea 以上のエネルギーを持つ割合が exp(−Ea/RT) で決まる」という統計力学の結論。シミュレーションでもちゃんとそうなる、というのを自分の目で確認できました。
自由研究の提案
このシミュレーションは、ちょっといじるだけで色んな探究テーマに化けます。
① 触媒の効果: 「触媒がある場所では Ea が下がる」というルールを加えてみる。同じ温度で反応速度がどう変わるか。 ② 可逆反応: AB → A + B の逆反応を入れて、平衡状態(A と AB の濃度が一定になる)を観察。 ③ 連鎖反応: A + B → AB、AB + C → ABC のように複数段の反応を組み合わせる。律速段階が見えてきます。 ④ 濃度効果: N_A, N_B を変えて、反応速度が「濃度の何乗」に比例するか測る(反応次数の決定)。
次回予告
EP.12 は自分の研究を LaTeX で論文化。今回のような自由研究を、本物の論文形式で書き上げる方法。Overleaf を使えば、ブラウザだけで数式・図表入りの が出来ます。探究学習・大学入試小論文・将来の卒論まで一気に役立つツールです。
この記事の感想を教えてください
あなたの 1 クリックで、本当にこの記事は更新されます。「もっと詳しく」「続編希望」が一定数集まった記事は、 ふくふくが 実際に内容を拡充したり続編記事を公開 します。 送信したリアクションはお使いのブラウザに記録され、再カウントされません。